
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
SCHEDULE
14A
(Rule 14a-101)
INFORMATION REQUIRED IN PROXY STATEMENT
SCHEDULE 14A INFORMATION
Proxy Statement Pursuant to Section 14(a) of The Securities Exchange Act of 1934
(Amendment No. )
Filed by the Registrant ¨
Filed by a Party other than the Registrant x
Check the appropriate box:
| ¨ | Preliminary Proxy Statement |
| ¨ | Confidential, for Use of the Commission Only (as permitted by Rule 14a-6(e)(2)) |
| ¨ | Definitive Proxy Statement |
| x | Definitive Additional Materials |
| ¨ | Soliciting Material Under Rule 14a-12 |
CytoDyn Inc.
(Name of Registrant as Specified in Its Charter)
PAUL A. ROSENBAUM
JEFFREY PAUL BEATY
ARTHUR L. WILMES
THOMAS J. ERRICO, M.D.
BRUCE PATTERSON, M.D.
PETER STAATS, M.D., MBA
MELISSA YEAGER
CCTV PROXY GROUP, LLC
(Name of Persons(s) Filing Proxy Statement, if Other Than the Registrant)
Payment of Filing Fee (Check the appropriate box):
| x | No fee required. |
| ¨ | Fee computed on table below per Exchange Act Rules 14a-6(i)(1) and 0-11. | |
| (1) | Title of each class of securities to which transaction applies: | |
| (2) | Aggregate number of securities to which transaction applies: | |
| (3) | Per unit price or other underlying value of transaction computed pursuant to Exchange Act Rule 0-11 (set forth the amount on which the filing fee is calculated and state how it was determined): | |
| (4) | Proposed maximum aggregate value of transaction: | |
| (5) | Total fee paid: | |
| ¨ | Fee paid previously with preliminary materials: | |
¨ Check box if any part of the fee is offset as provided by Exchange Act Rule 0-11(a)(2) and identify the filing for which the offsetting fee was paid previously. Identify the previous filing by registration statement number, or the form or schedule and the date of its filing.
| (1) | Amount previously paid: | |
| (2) | Form, Schedule or Registration Statement No.: | |
| (3) | Filing Party: | |
| (4) | Date Filed: | |
The participants named herein (collectively, the “Participants”), have filed a definitive proxy statement and accompanying WHITE proxy card with the Securities and Exchange Commission to be used to solicit votes for the election of its slate of director nominees at the 2021 annual meeting of stockholders of CytoDyn Inc., a Delaware corporation (the “Company”).
On September 24, 2021 the Participants issued a press release, a copy of which is filed as Exhibit 1 hereto, announcing their strategic plan to obtain cancer therapy approval for Leronlimab. The press release contains a link to the full plan, a copy of which is filed as Exhibit 2 hereto. The Participants also posted the press release and the full plan to their website at www.advancingll.com and e-mailed them to certain stockholders of the Company.
Exhibit 1—Press Release dated September 24, 2021
GROUP OF CYTODYN STOCKHOLDERS Shares strategic plan to obtain cancer therapy approval for leronlimab
Plan Designed to Generate Much-Needed Revenue and Receive FDA Approval for Currently Mismanaged Drug
Believes Company Cannot Continue to Pursue Status Quo Strategy, Which Has Been Abjectly Discredited
NEW YORK — September 24, 2021 — A group of long-time stockholders (the “Nominating Stockholders” or the “Group”) of CytoDyn Inc. (“CYDY or the “Company”) (OTC: CYDY) that has nominated five highly experienced director candidates to serve on the Company’s Board of Directors today announced its comprehensive strategic plan to obtain cancer therapy approval for Leronlimab. If implemented, the Group believes this plan would begin to generate much-needed revenue for CYDY and position the Company to earn FDA approval for the drug, while enhancing the value of all stockholders’ shares.
The Group’s strategy is designed to overcome current CYDY management’s failures to implement a coherent, effective plan to generate revenue and obtain FDA approval for Leronlimab. While the Group has tried to discuss the enormous potential of its oncological strategy to reinvigorate CYDY with the Board and management team, they have refused to meaningfully engage every step of the way. Instead, incumbent Company leadership has decided to focus on entrenching themselves and clinging to their outsized compensation packages at the expense of shareholders.
The Group’s approach to cancer therapy will be scientifically valid and extremely efficient and will be critical to unleashing the potential value of Leronlimab and the investments of all stockholders. The full plan can be found at here, and highlights are as follows:
| · | Utilize real data based on precision medicine determination of Leronlimab binding cancers; |
| · | Apply both combination therapy with complementary immune-oncology blockbuster drugs and adjuvant monotherapy in CCR5+ tumors; |
| · | Prioritize cancer targets based on CCR5 expression; and |
| · | Partner with leading oncology companies that lack a CCR5 asset like Leronlimab. |
Dr. Bruce Patterson, one of the Group’s five nominees and a renowned virologist, pathologist, and cancer technology pioneer, stated: “We are excited to present our plan on how to maximize the value of Leronlimab and save the lives of countless cancer patients. We believe we have put in place a strategy that is not only executable but also best positions CYDY for future success and will repair the Company’s standing among the medical, regulatory, and investment communities. In order to protect the value of your investment in CYDY, we strongly you recommend you for our highly qualified director nominees on the WHITE proxy card today.”
Dr. Patterson added, “Immuno-oncology (IO) has revolutionized oncological therapy for many cancers and makes up one of the largest cancer drug market segments. However, CYDY’s Board and management have utterly failed to capitalize on this tremendous opportunity and have not publicly presented any strategic plan on how they might make Leronlimab a reality. On the other hand, our cancer program will combine the value that Leronlimab has as a potential adjuvant therapy following treatment with the urgent opportunities that exist in IO and in doing so, will create enormous value for CYDY and its stockholders. This carefully designed plan is critical to the Company’s success, and we are excited by the prospect of finally being able to implement it.”
The full text of the Group’s cancer therapy approval plan can
be accessed at:
www.advancingll.com/cancerplan
All CYDY shareholders are reminded that your vote is essential to charting a course towards lasting value creation and holding the current Board and management team accountable for the immense value destruction they have overseen throughout their tenure. Help us enable CYDY to achieve its incredible potential by voting the WHITE proxy card to elect the Group’s five independent director nominees today.
Important Information
Paul Rosenbaum, Jeffrey Beaty, Arthur Wilmes, Thomas Errico, M.D., Bruce Patterson, M.D., Peter Staats, M.D., Melissa Yeager and CCTV Proxy Group, LLC (collectively the “Participants”) have filed a definitive proxy statement and accompanying WHITE proxy card with the Securities and Exchange Commission (the “SEC”) to be used in connection with the solicitation of proxies from the stockholders of CytoDyn Inc. (the “Company”). All stockholders are advised to read the definitive proxy statement and other documents related to the solicitation of proxies. The definitive proxy statement and an accompanying proxy card is available at no charge on the SEC’s website at http://www.sec.gov/. In addition, the Participants will provide copies of the proxy statement, without charge, upon request. Requests for copies should be directed to the Participants’ Proxy Solicitor, Okapi Partners LLC, by calling (844) 202-7428.
Disclaimer
This material does not constitute an offer to sell or a solicitation of an offer to buy any of the securities described herein in any jurisdiction to any person. In addition, the discussions and opinions in this press release and the material contained herein are for general information only and are not intended to provide investment advice. All statements contained in this press release that are not clearly historical in nature or that depend on future events are “forward-looking statements,” which are not guarantees of future performance or results, and the words “anticipate,” “believe,” “expect,” “may,” “could,” and similar expressions are generally intended to identify forward-looking statements. Forward looking statements contained in this release are based on current expectations, speak only as of the date of this press release and involve risks that may cause the actual results to be materially different. Certain information included in this material is based on data obtained from sources considered to be reliable. No representation is made with respect to the accuracy or completeness of such data. The Participants disclaim any obligation to update the information herein and reserve the right to change any of their opinions expressed herein at any time as it deems appropriate.
Contacts:
Media
Mark Semer/Sam Cohen
Gasthalter & Co.
(212) 257-4170
cydy@gasthalter.com
Investors
Bruce Goldfarb/Chuck Garske
Okapi Partners
(212) 297-0720
info@okapipartners.com
Exhibit 2—Strategic Plan to Obtain Cancer Therapy Approval for Leronlimab
Cancer Product Development Plan: Leronlimab
EXECUTIVE SUMMARY
This document provides an overview of the Cancer Product Development Plan tendered by the Board slate proposed by nominating shareholders Jeff Beaty, Paul Rosenbaum, and Arthur Wilmes. The Board slate has not seen a cancer plan proffered by CytoDyn, Inc. (CYDY or the Company). As such, this plan may draw inferences from public statements made about and actions taken by the Company as it regards cancer studies and potential clinical trials. Our intent is to incorporate the findings from previous company studies when given the opportunity to review the details of the studies and the data collected from such studies.
The following is a high-level summary of the Cancer Product Development Plan:
| 1. | General Direction. We differ from the Company as to the general direction of proposed cancer targets and clinical trial approaches to a cancer therapy approval. |
| 2. | Combination Therapy Versus Monotherapy. We believe that the Company’s best opportunity for cancer approval is with large pharmaceutical company partners and in combination trials with a class of drugs called PD-L1/PD-1 inhibitors. |
| 3. | Immune Checkpoint Inhibitors (ICI) Alternatives. We believe that Leronlimab's safety record will make it a preferred candidate as an adjunct ICI treatment alternative to be combined with PD-L1/PD-1 inhibitors. |
| 4. | Pharmaceutical Company Partners. We will focus discussions on four specific pharmaceutical company partnership candidates: Roche- TECENTRIQ® (Atezolizumab), AstraZeneca- IMFINZI® (Durvalumab), Pfizer/EMD Serono - BAVENCIO® (Avelumab), and Regeneron- LIBTAYO® (Cemiplimab). These companies are prospective targets for business reasons such as expiring exclusivity or they do not have CCR5 assets as combination therapies. |
| 5. | Cancer Targets. Cancer targets will be defined by the IncellDx H-Scoring system. (The H-Scoring system will be licensed from IncellDx to assist in assessment of cancer opportunities.) Potential initial targets include Lung Adenocarcinoma, Lung Squamous Cell Carcinoma (SCC), Breast Cancer, Pancreatic Cancer, Urothelial Cancer, and Colorectal Cancer. |
| 6. | Clinical Research Organization (CRO). The Company had used Amarex as its CRO for previous cancer related studies. Amarex does not have oncology trial management experience and did not employ medical oncology physicians in the conduct of prior studies. We will be using a highly qualified CRO, PI, and professionals experienced in the administration of PD-L1/PD-1 inhibitors. |
| 7. | Financing. Contingent upon successful completion of a long-hauler study and approval by the FDA. We can then make a compelling scientific safety and efficacy case to prospective pharma partners. We believe significant support from a pharmaceutical company partner will reduce overall cost. Also, we believe having a significant partner, along with a win with long hauler use approval, will be our value proposition for financial support. |
| 8. | Timing. The planning and expanded cancer plan will be developed coterminous with the long hauler study. The lead time for a cancer trial will be much longer due to all the research and planning required. This includes, but is not exclusive to, Leronlimab safety and efficacy data (and FDA approval), initial partner presentations and discussions, contract negotiations, FDA meetings, and other requirements in advance of trial enrollment. |
| 1 | Page |
Cancer Product Development Plan: Leronlimab
PRE-LAUNCH ANALYSIS
Cancer Therapy Opportunities for Leronlimab
Opportunity: Combination therapy with PD-L1 Inhibitors
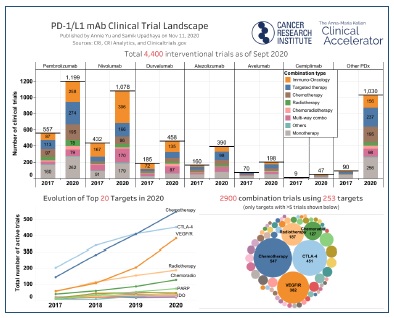
The treatment of cancer has been profoundly changed with the use of immune-oncology drugs such as the PD-L1 inhibitors: (1) OPDIVO®, (2) KEYTRUDA®, and (3) TECENTRIQ®1. Not only have these drugs proven to be successful in multiple tumor types but they also represent some of the most successful drugs in the market in terms of revenue.
PD-L1/PD-1 inhibitors act to re-engage the immune cells, in the tumor microenvironment, against the tumor. Eventually this response is tempered by immune exhaustion and infiltration of the tumor microenvironment by T-regulatory cells. Recent studies are showing that the response to PD-L1/PD-1 inhibitors, Immune Control Point (ICI) inhibitors, is controlled by inhibitory mechanisms that the body uses to prevent autoimmunity. Pharma has now found ways to extend the anti-tumor activity of these inhibitors by using combination therapies that inhibit the inhibitory mechanisms thereby extending the enhanced immune response against tumors from PD-L1/PD-1 inhibitors.
Combination Therapies
Opportunity: Leronlimab should prove to be a superior ICI treatment alternative.
Pharma has developed drugs to be used in tandem with the PD-L1/PD-1 inhibitors to “release the brake” they applied to these blockbuster drugs. Ipilimumab (CTLA-1)2 and relatlimab (Lag-3)3 act by inhibiting T-effector cell exhaustion and by inhibiting T-regulatory cells. Current available monoclonal antibody medications (MABs) ipilimumab (Yervoy) and relatlimab (Lag-3) are being used in combination with PD-L1/PD-1 inhibitors to facilitate the inhibitory mechanisms. These drugs have large market potential as Yervoy already generates $1.5 billion in revenue, expected to exceed $2 billion in annual revenue by 20264.
1 Immuno-oncology drugs that block the checkpoint PD-1 have been one of the biggest success stories in the pharma industry, creating blockbusters like Bristol Myers Squibb’s Opdivo and Merck’s Keytruda. The embrace of the drugs to treat many types of metastatic cancer has created a $23 billion market. New use applications will increase projected revenue as use in “pan-adjuvant” settings, in which patients are given PD-1 blockers before and/or after surgery along with other treatments to boost their chances of beating their cancer. Market analyst, Bernstein Research, said in a note to investors earlier this week that, for Merck and BMS, pan-adjuvant approvals could add at least $12 billion to their annual revenue from PD-1 drugs in the next eight years—with the lion’s share of that going to Keytruda. (Fierce Pharma: How can the $23B market for PD-1 blockers grow? 'Pan-adjuvant' use is key: report; Arlene Weintraub | Dec 3, 2020.
2 Tarhini A, Lo E, Minor DR. Releasing the brake on the immune system: ipilimumab in melanoma and other tumors. Cancer Biother Radiopharm. 2010;25(6):601-613. doi:10.1089/cbr.2010.0865
3 Lipson EJ, Tawbi HAH, Schadendorf K, et al: Relatlimab plus nivolumab versus nivolumab in first-line advanced melanoma: Primary phase III results from RELATIVITY-047 (CA224-047). 2021 ASCO Annual Meeting. Abstract 9503. Presented June 6, 2021.
4 Forecasted sales of Yervoy worldwide from 2020 to 2026, by cancer type (in million U.S. dollars); Statista
| 2 | Page |
Cancer Product Development Plan: Leronlimab
Combination therapy clinical trials have become the predominant trial form in oncology5.
Checkpoint inhibitor therapy has introduced a revolution in contemporary anticancer therapy. It has led to dramatic improvements in patient outcomes and has spawned tremendous research into novel immunomodulatory agents and combination therapy that has changed the trajectory of cancer care6. ICI inhibitors represent a significant advance in the management and survival of cancers such as melanoma or non-small cell bronchial carcinoma, however, they can induce unusual side effects, such as hypophysitis7 resulting in pituitary gland enlargement and pituitary/hypothalamic dysfunction. A study of patients receiving ICI inhibitors found elevated risk of hypophysitis. In all cases, no patient recovered their previous hormonal function. The mean time of onset was significantly shorter with ipilimumab than other ICIs. ICI-related hypophysitis generate deficits that do not spontaneously recover, even at a distance from the event, meaning patients will require long-term coordinated onco-endocrinological management8.
Leronlimab would perform the same function as combination ICI inhibitors by inhibiting T-regulatory cell migration, repolarizing macrophages from M2 (pro-inflammatory/pro-tumor) to M1 (effector, anti-tumor). Evidence found in the HIV and Covid trials indicated that Leronlimab reversed T-cell exhaustion, and also increased Granzyme A (the cancer bullets) in CD8 T-cells.
5 Nature Reviews Drug Discovery; March 2021 volume 20 no. 3 www.nature.com/nrd
6 Hermel DJ, Sigal D. The Emerging Role of Checkpoint Inhibition in Microsatellite Stable Colorectal Cancer. J Pers Med. 2019;9(1):5. Published 2019 Jan 16. doi:10.3390/jpm9010005
7 Faje, A. Hypophysitis: Evaluation and Management. Clin Diabetes Endocrinol 2, 15 (2016). https://doi.org/10.1186/s40842-016-0034-8
8 Garon-Czmil, J., Petitpain, N., Rouby, F. et al. Immune check point inhibitors-induced hypophysitis: a retrospective analysis of the French Pharmacovigilance database. Sci Rep 9, 19419 (2019). https://doi.org/10.1038/s41598-019-56026-5
| 3 | Page |
Cancer Product Development Plan: Leronlimab
Leronlimab has an opportunity to be a major replacement option for ICI’s due to toxicity concerns. Such toxic side effects are major disorders requiring lifelong drug replacement therapies such as insulin for Type-1 DM. Patients developing panhypopituitarism have a life-threatening illness that will be extremely expensive to treat. Leronlimab’s safety profile, as indicated by HIV and Covid trial results, would reduce the toxicity risk. Payer, FDA, and Medicare communications will be able to show economic value associated with the reduction in pituitary disorders, reduced organ dysfunction (e.g., nephritis, hepatitis, etc.), lower toxicity side effect costs to treat, and improved quality of life.
Involvement of the CCL5/CCR5 Axis in Cancer Progression/Tumors With the Highest Expression Levels of CCR5
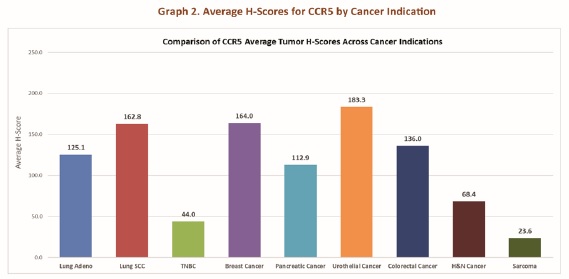
Opportunity: Lung Adenocarcinoma, Lung Squamous Cell Carcinoma (SCC), Breast Cancer, Pancreatic Cancer, Urothelial Cancer, and Colorectal Cancer have the highest H-Scores and are potential priority cancer clinical trial targets.
CCR5 engagement increases tumor growth in both autocrine and paracrine manners. Autocrine growth is characterized by the presence of functional CCR5 and the secretion of its ligands by cancer cells, while paracrine growth is induced by CCR5 ligands secreted by normal cells of the TME-like T lymphocytes, macrophages, fibroblasts, MSCs and CAFs. CCR5 ligands stimulate cell growth by inducing the mammalian target of rapamycin (mTOR) pathway followed by a rapid up-regulation of cyclin D1, c-Myc and Dad-1 expression, by activating the Jak–STAT pathway, and by enhancing glucose uptake with increased ATP production and glycolysis associated with extracellular acidification.9
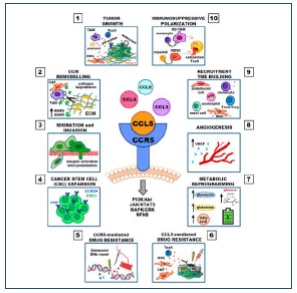
The CCL5/CCR5 axis exerts profound effects on the tumor progression in several hematological cancers. CCR5 expression is highly variable on immune cells and certainly on tumors, however, CCR5 expression is highly regulated by inflammation especially as displayed by certain cytokines10. A quantitative, CCR5 immunohistochemistry (IHC) assay from IncellDx can be used to create a reproducible scoring system (H-score) to assess the level of CCR5 expression. (The H-Scoring system will be licensed from IncellDx to assist in assessment of cancer opportunities.) The H-Scoring system will be used to dictate the hematological cancers to initially be pursued as top opportunities for Leronlimab clinical trials and combination therapies. Cancers with the highest CCR5 expression are priority candidates for further analysis and clinical trial engagement.
9 The CCL5/CCR5 Axis in Cancer Progression; Donatella Aldinucci *, Cinzia Borghese and Naike Casagrande
10 Patterson, B., Czerniewski, M.A., Andersson, J., Bauer, Y., Su, F., Jiyamapa, D., Burki, Z., Landay, A. Regulation of CCR5 and CXCR4 expression by Type 1 and Type 2 cytokines: Type 1 cytokine upregulation of CCR5 expression can be abolished by IL-10. Clin Immunol 1999 91:254-262
| 4 | Page |
Cancer Product Development Plan: Leronlimab
Partnership Opportunities
Opportunity: Bristol-Myers Squibb, Roche, Astra Zeneca, Pfizer/Serono, and Regeneron are primary targets for combination trial negotiations.
OPDIVO® (nivolumab) is losing patent protection11 (Europe will expire in 2026, followed by that in the US in 2027) and will increase the number of companies in the immune-oncology space but more importantly will increase the number of companies with PD-1 antagonists that do not have CCR5 assets as combination therapies. Large Pharma companies with PD-L1/PD-1 inhibitors with no CCR5 assets include:
| 1. | Roche- TECENTRIQ® (Atezolizumab) |
| 2. | Astra Zeneca- IMFINZI® (Durvalumab) |
| 3. | Pfizer/EMD Serono - BAVENCIO® (Avelumab) |
| 4. | Regeneron- LIBTAYO® (Cemiplimab) |
These companies should be amenable to clinical investigation into Leronlimab combination once Leronlimab proves its efficacy as a CCR5 inhibitor.
INVESTIGATION STAGE
FDA Approval Planning
Process Step: Schedule an INTERACT meeting with CBER/FDA as soon as the Company formulates a product-derivation strategy to evaluate in a clinical study.
The pre-launch analysis will result in an internal strategy to evaluate in a clinical study. A meeting with the FDA will be scheduled as soon as the analysis is completed. This is a primary condition for requesting an Initial Targeted Engagement for Regulatory Advice on Products (INTERACT) meeting. An INTERACT meeting is an informal non-binding consultation with the Center for Biologics Evaluation and Research (CBER) at FDA12.
11 Pairing with Leronlimab in combination therapy could increase longevity.
12 SOPP 8214: INTERACT Meetings with Sponsors for Drugs and Biological Products
| 5 | Page |
Cancer Product Development Plan: Leronlimab
Our evaluation of the CBER and FDA is that CCR5 inhibitors are still a relatively new class of drugs for them. As such, it is incumbent upon the company to have face-to-face meetings with the CBER/FDA to present the clinical science, diagnostic testing, and general patient administration processes for Leronlimab. An INTERACT meeting benefits both the company and CBER/FDA. The company will be able to receive initial, non-binding advice from FDA regarding chemistry, manufacturing and controls (CMC), pharmacology/toxicology, and/or clinical aspects of the development program.
The Company’s relationship with the CBER/FDA will allow the Company to “get off on the right foot” with the regulatory bodies. The Company is engaging the CBER/FDA in the entire clinical trial development process. This meeting with benefit the Company and (1) assist it in conducting early product characterization and preclinical proof-of-concept studies, (2) initiate discussion for a novel drug therapy, (3) create a collaboration between the Company and CBER/FDA as to overall early-phase clinical trial design elements, and (4) identify critical issues or deficiencies among both parties that need to be addressed as part of the drug evaluation process.
Principal Vendor Selections
Process Step: Critical vendor selection and the negotiation of contracts: Master Services Agreement, Statement of Work, and Ownership of Intellectual Property and Data.
Clinical Research Organization: The Clinical Research Organization (CRO) will be responsible for managing the clinical trial process. The CRO will be recruited from among a pool of candidates with advanced experience with and knowledge of oncology and hematology clinical trial design, process, and administration.
Principal Investigator: The Principal Investigator (PI) will be responsible for leading the design and conduct of the clinical trial. The PI will be recruited from among a pool of candidates with advanced experience with and knowledge of immune-oncology drugs such as the PD-L1 inhibitors and the molecular testing that optimizes the administration of combination ICI inhibitors. The PI, in consultation with the Company, will recruit Clinical Investigations Leaders and Program Leaders.
Molecular Diagnostics: A Molecular Diagnostics vendor (MDV) will be responsible for evaluating the metabolic and pharmacologic effects and clinical outcomes associated with treatments in the trial. The qualifications for an MDV will include vendors with experience with and knowledge of the molecular and cell biology of cancer to improve diagnosis and therapy. The MDV will be tasked with the identification and assessment of five clinical markers: (1) Pharmacokinetic, (2) Pharmacodynamic, (3) Prognostic, (4) Predictive, and (5) Pharmacogenetic markers. These markers are expected to be a primary topic of discussion with CBER/FDA during the INTERACT meeting. The MDV will be responsible for enforcing the rigor of the clinical trial as the markers will inform (1) patient eligibility for the trial, (2) patient assignment to therapy, dose selection, and (3) patient stratification within a trial.
| 6 | Page |
Cancer Product Development Plan: Leronlimab
Supply Chain Management Plan
Process Step: Begin planning the supply chain management strategy.
The supply chain management process entails a number of steps as follows:
| 1. | Negotiate contract terms with primary drug distributors. AmerisourceBergen, Cardinal Health, and McKesson manage approximately eighty-five%13 of all drug distribution in the US and will be the initial partner targets. |
| 2. | Estimate patient demand for efficient distributor supply management. Create a pricing strategy based upon expected demand, future competition, and projected marketing cost. Define the wholesale acquisition cost (WAC). |
| 3. | Create the payer contracting strategy. This includes primary insurance companies (and managed care organizations), pharmacy benefit managers, government payers (Medicare and Medicaid), and large health systems. Supplement strategies with managed market value propositions to support negotiations. |
Clinical Trial
Process Step: Clinical trial development is a collaborative effort among the collaboration partner, the Company, CDM, MDV, and regulatory agencies.
This section is a broad outline of the clinical trial process. A more detailed plan will be created after selection of the PI, CRO, and MDV. The clinical trial protocol design will reflect discussions and findings from the initial INTERACT meeting. The trial process will follow these general steps:
| 1. | Clinical Trial Collaboration Agreement (CTCA). Negotiate the CTCA ensuring a clear definition of ownership rights, patent protections, data ownership, statement of work, sponsor versus supporting roles/responsibilities, supply obligations, intellectual property ownership, and other critical elements intended to promote a collaborative effort while maintaining ownership of critical company intellectual property. |
| 2. | Partner Collaboration. The combination study partner will have more experience than the Company with regard to the appropriate oncology protocols and will heavily influence trial design. |
| 3. | Data Management Strategy. Clinical Data Management (CDM) is a critical element of the trial and will require a separately designed CDM protocol. A well designed CDM protocol can lead to faster commercialization. technology. The CDM protocol will also be integrated into the trial protocol. The CDM must be compliant with all relevant CDM society protocols for clinical research. The general CDM work plan begins at trial inception. Critical elements include, but not exclusively, (1) data quality standards, (2) case report form (CRF) design, (3) CRF annotation requirements, (4) database design, (5) data-entry standards, (6) data validation protocols, (7) conflict resolution process for data discrepancies, (8) medical coding protocols, (9) data extraction protocols, and (10) database security protocols. |
| 4. | MDV Integration. The INTERACT meeting will have been the basis for the MDV protocol. The MDV protocol is integrated into the trial protocol. Critical elements of the MDV protocol include, but not exclusively, the following items: (1) safety and efficacy marker identification, (2) frequency of testing/assessment, (3) how test results will be reflected in decision making, (4) relationship between test results and dosing, and (5) data reporting that reflects the efficacy demonstration. Most importantly, the team must all agree to the correct biomarkers necessary for trial success. |
| 5. | Regulatory Relationships. Prior meetings with the CBER/FDA will guide the process of interim reporting and consultations. |
| 6. | Trial Design. This will be a holistic process led by the PI with collaboration among the PI, partner company, CRO, trial managers, MDV, and CDM. The goals of the trial should result directly from all the preliminary discussions and educations sessions with regulatory agencies. Critical elements include, but not exclusive to, the following: (1) group size required for statistical power, (2) randomization procedures, (3) stratification of randomized patient groups, (4) eligibility criteria and determination methods, (5) protocol adherence and variation controls, (6) define Type I and Type II errors, (7) dealing with missing data, (8) interim reporting, (9) endpoints, (10) placebo treatment and control group definition, and (11) other critical criteria intended to best assess and differentiate the efficacy of the test drug to placebo. |
13 Financing and Distribution of Pharmaceuticals in the United States; Matan C. Dabora, MD, MBA1; Namrata Turaga, MD, MBA1; Kevin A. Schulman, MD, JAMA. 2017;318(1):21-22. doi:10.1001/jama.2017.5607
| 7 | Page |
Cancer Product Development Plan: Leronlimab
POST-TRIAL ANALYSIS
Process Step: Post-Trial analysis plan will be required for FDA surveillance.
Post-Market Surveillance
Post-market surveillance is the requisite monitoring of the drug combination safety once introduced into the market. The purpose is to identify previously unrecognized adverse and positive effects. This step will require a separate protocol including, but not exclusive to, the following items: (1) data collection, (2) indicator criteria to assess statistically credible new effects (adverse and positive), (3) cost-benefit analysis and interim calculational frequency, (4) and compliance with all FDA post-market reporting requirements.
Evidence Development
Post-Trial analysis evidence gathering can support further clinical research, clinical trials, and additional collaboration opportunities.